Structure and Function of Telomeres and Telomerase


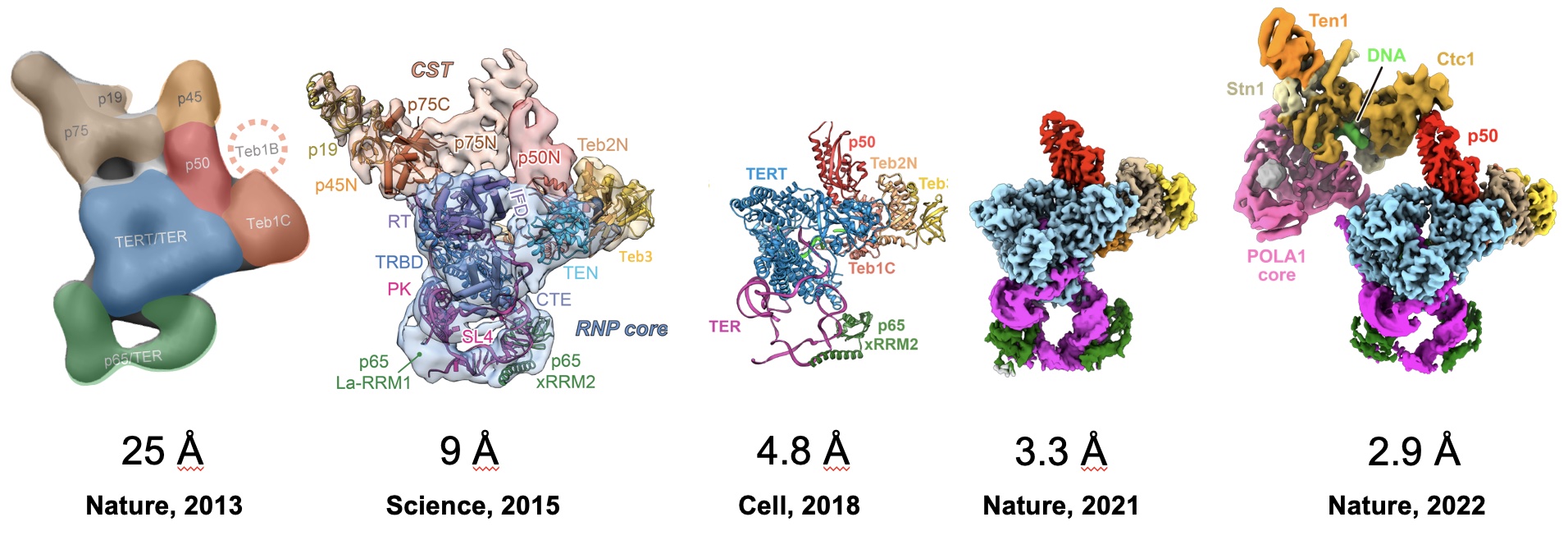
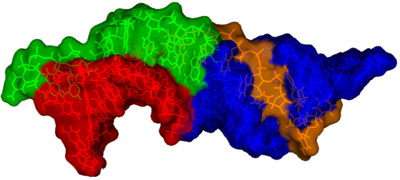
Tetrahymena telomerase A current major focus of the lab is the study of telomerase structure and function. The telomere repeat sequence is not replicated by the normal DNA replication machinery, but rather by a unique RNA-protein enzyme complex called telomerase. Telomerase uses an RNA template that is an integral part of the enzyme and a specialized reverse transcriptase (TERT) to processively synthesize the G-rich strand. Telomerase has only a low or undetectable level of activity in normal somatic cells, but is highly active in most (>90%) cancers, and is thus of interest as a target for anticancer drugs. In addition, mutations in both the telomerase RNA and telomerase proteins are associated with some diseases of the haemopoeitic system such as some forms dyskeratosis congenita and aplastic anemia. In collaboration with Kathleen Collins's laboratory at Berkeley and Hong Zhou's Laboratory at UCLA, we recently determined the architecture of the complete Tetrahymena telomerase holoenzyme by electron microscopy.
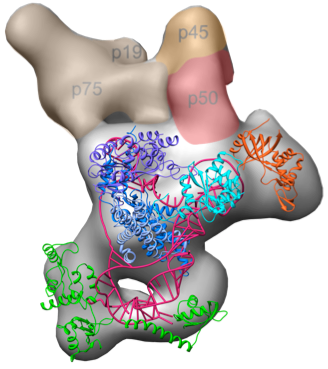
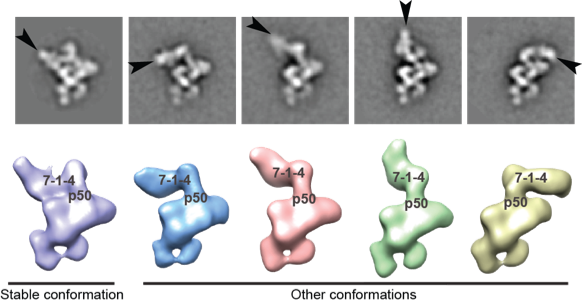
In addition to solving the complete structure by EM, we were able to localize each subunit by antibody and viral coat protein binding. The added density from the bound proteins effectively “pinpointed” the subunits, allowing us to use the contours seen by EM to assign each subunit. Two surprising discoveries from this project include the central role p50 plays—previously thought to be substoichimetric—and that the p75-p19-p45 subcomplex rotates as an intact subunit. Read more ...
Human telomerase RNA Human telomerase contains a 451 nt RNA along with a variety of proteins besides the telomerase reverse transcriptase. Only about 10 nt are needed for the template, so what is the function of all that extra RNA? This is one of the major questions that we trying to answer, by investigating the structure of various domains of the telomerase RNA andtheir interactions with telomerase proteins. Within the telomerase RNA there are domains that are essential, together with TERT, for catalysis as well as domains required for telomerase RNA accumulation, localization, and 3’ end processing. Our laboratory has determined structures of essential domains of telomerase RNA, providing the first structural insights into the roles of these domains in telomerase biology.
Most recently, we have investigated the structure and dynamics of the two helical regions of the pseudoknot/core domain, P2ab which contains a 5 nucleotide bulge and P2a.1 which is a mammalian specific extension which contains an asymmetric internal loop. We determined the NMR structure and investigated the dynamics P2ab and found that the 5 nt bulge (J2a/b) forms a defined S-shape and creates an ~90° bend between flanking helices that has a surprisingly limited inter-helical flexibility. Nucleotide substitutions in J2a/b that affect the bend angle, direction, and inter-helical dynamics are correlated with telomerase activity. We also developed a new combined modeling and NMR approach (RDC-MC-Sym approach) and used it to model the structure of the remaining helical sub-domain of the core domain that is too conformationally flexible to be determined by NMR alone. We combined the three sub-domain structures to determine the first useful model of the human telomerase RNA core domain.The model and dynamics analysis show that J2a/b serves as the dominant structural element in defining the overall topology of the core domain, and suggest that inter-helical motions in P2ab facilitate nucleotide addition along the template and template translocation. In addition, we found that the J2a/b bulge belongs to a rare 5 nucleotide bulge family that has a conserved structure. The only other example of a 5 nucleotide bulge in the PDB is found in domain II of HCV IRES, where its bend is also proposed to be functionally important. Our ongoing work and long term goal is aimed at complete structure determination of the catalytic core of human telomerase, including the other regions of the telomerase RNA essential for catalysis and the telomerase reverse transcriptase. Read more ...
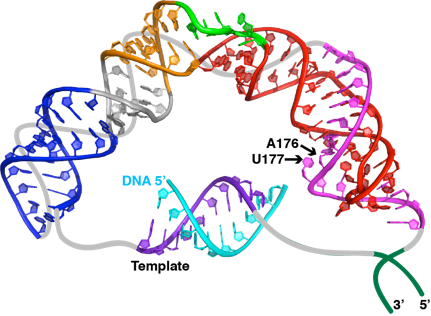
Localization, accumulation, and 3’ end processing The 3’ end of vertebrate telomerase RNA contains a domain identified by the Collins laboratory as an H/ACA RNA. H/ACA RNAs, together with the H/ACA proteins, form an RNP that generally functions to convert specific Us in mRNA or snRNA to the modified nucleotide pseudouridine. While there does not appear to be any role in pseudouridylation for the H/ACA domain of vertebrate telomerase, its 3’ terminal hairpin loop, called the CR7 domain, had been proposed to be essential for localization, accumulation, and 3’ end processing of the telomerase RNA. Further, it had been proposed that the loop contained a sequence that targeted telomerase to Cajal bodies (making it an H/ACA scaRNA) rather than to the nucleolus (an H/ACA snoRNA) and an uncharacterized potentially telomerase RNA 3’ end processing signal.
To dissect out the loop contributions to both processing and localization, we determined the solution structures of three hairpin loops, the CR7 terminal loop of hTR, the 3’ terminal loop of the human U64 H/ACA snoRNA, and the 5’ terminal loop in the H/ACA domain of the human U85 C/D-H/ACA scaRNA. Based on comparison of the structures, we designed mutant hairpin loops and investigated the effects of these nucleotide substitutions of telomerase RNA localization and processing in vivo using FISH (fluorescence in situ hybridization) and RNAse A/T1 mapping, respectively (collaboration with Kiss laboratory). Comparison of the structures, together with results of the localization and processing studies of the wt and mutant full length and 3’ terminal H/ACA scaRNA domain of human telomerase RNA constructs, revealed the structural elements of the Cajal body localization signal and identified the sequence and structural requirements of the hTR processing signal. We further found that these two signals are functionally independent. These studies also revealed that in the absence of processing the telomerase RNA remains at the transcription site in nucleoplasmic foci, indicating that 3’ end processing is a prerequisite for localization. Read more ...
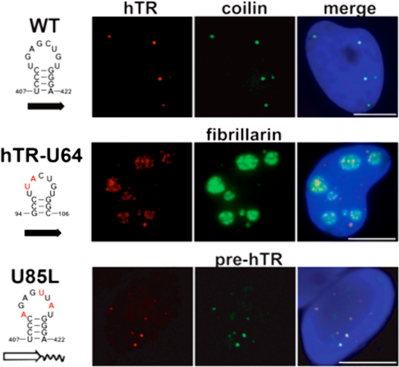
A growing body of experimental evidence indicates that efficient accumulation and proper subnuclear localization of human telomerase RNA is fundamental to the correct function of human telomerase. Therefore, our results have facilitated the understanding of the regulation of human telomere synthesis. As an example, our structural and functional studies of the CR7 domain also provided an explanation for how mutation associated with dyskeratosis congenita in CR7 affects telomerase. Although this mutation is in the upper stem of the CR7 domain, we found that it results in a rearrangement of the CR7 loop that affects the processing signal. We showed that the mutation abolishes hTR processing, and that a compensatory mutation that restores correct base pairing in the stem results in correct processing and localization to Cajal bodies.
Riboswitch Structure and Dynamics
A major new area of interest in the laboratory is investigations into riboswitch structure and function. Riboswitches are a recently discovered class of RNA elements that function as an important regulatory mechanism for control of gene expression in almost all classes of prokaryotes (bacterial and archaea), as well as in some eukaryotes such as algae. Like other classes of RNA that have recently been found to have fundamental roles in controlling gene expression, such as siRNAs, their existence and prevalence have only recently been appreciated. Nevertheless, riboswitches help regulate more than 150 genes in B. subtilis genome alone, and an understanding of their function is central to understanding gene regulation. The riboswitch RNA sequences, which are usually located in the 5’ untranslated region of operons, are unique in requiring no auxiliary protein factors. Rather, binding of a specific metabolite induces a conformational change in the RNA, resulting in repression or activation of transcription, translation, or RNA processing of the associated genes. Currently, at least twenty different classes of riboswitches have been identified, which bind ligands that include purine nucleobases, vitamin cofactors, amino acids, metal ions, and even second messengers such as cyclic diGMP.
Riboswitches are typically composed of two domains: an “aptamer” domain that serves as the receptor for the metabolite and an “expression platform” whose secondary or tertiary structure signals the regulatory response. The metabolite binds to a 5’ aptamer domain in the riboswitch, generally resulting in sequestration or formation/exposure of a terminator or a ribosome binding sequence as the RNA is being transcribed or translated. Since the discovery of riboswitches, extensive efforts have been made in a number of laboratories to determine their structures in order to understand how they function to regulate gene expression. This has been very successful, with a large number of crystal structures of the aptamer domain with bound ligand determined. However, until our recent work, there have been no reported solution structures of riboswitches. Solution NMR studies offer the advantage of being able to look at not only the ligand-bound aptamer but also the ligand-free state, as well as RNAs that include the expression platform.
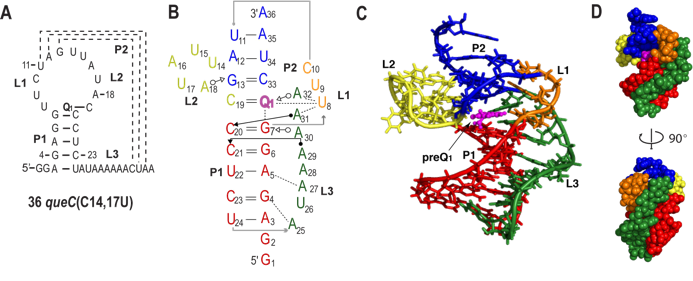
We have studied the preQ1 riboswitch. PreQ1 (7-amminomethyl-7-deazaguanine) is a biosynthetic precursor of queuosine (Q), a hypermodified guanine nucleotide found in the wobble position of GUN anticodons in tRNAHis, tRNAAsp, tRNAAsn, and tRNATyr. In eubacteria, free preQ1 is directly incorporated at the wobble position of the appropriate tRNA anticodon by a tRNA-guanine transglycosylase, replacing the unmodified G, and is subsequently further modified to yield queuosine. In general, modified nucleotides near the anticodon of tRNA are thought to modulate codon-anticodon interactions and affect translational fidelity, and Q has been implicated in a wide variety of cellular functions including eukaryotic cell proliferation and differentiation, tyrosine biosynthesis, and virulence in Shigella flexneri. The preQ1 riboswitch is unusual in that it contains the smallest known natural aptamer domain, consisting minimally of 34 nucleotides predicted to form a hairpin followed by a conserved A-rich ‘tail’. We addressed the question of how such a small aptamer domain could function in the riboswitch by solution NMR studies of the preQ1 riboswitch. Using state of the art NMR methods combined with structural analysis of ~20 mutant RNAs, we determined the solution structure of the preQ1 riboswitch aptamer from B. subtilis in complex with preQ1. The structure is a unique compact pseudoknot with three loops and two stems that encapsulates preQ1 at the junction between two stems. Despite its small size, the structure has a complex architecture that includes new and unusual RNA interactions. Our solution NMR studies revealed that folding of the preQ1 aptamer and formation of the binding pocket are completely dependent on concomitant preQ1 binding. Investigation of longer RNAs that include the regulatory regions showed that formation of the antiterminator precludes binding of preQ1 providing a model for how the riboswitch functions to regulate transcription. These structural studies of the smallest known natural riboswitch aptamer should help to guide the design of small and efficient artificial riboswitches. Another important aspect of this work is that the solution NMR studies showed that the binding pocket is surprisingly flexible and dynamic. This has led to a new area of investigation for the lab into the conformational dynamics of riboswitches. A crystal structure of the preQ1 riboswitch has also been reported, which is highly similar to the solution structure but has some differences in loop 1 and stem 2 that close the binding pocket. In ongoing work, we are using NMR methods to investigate how the ligand is ‘captured’ as the RNA is transcribed or translated. Read more ...
Biogenesis of H/ACA RNPs
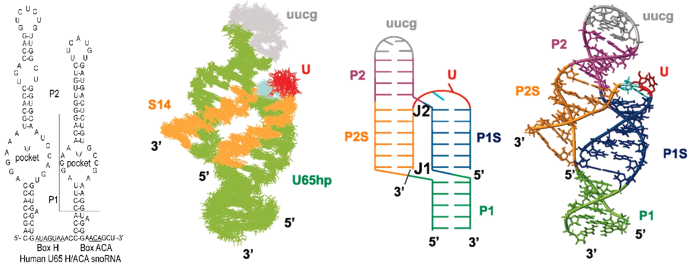
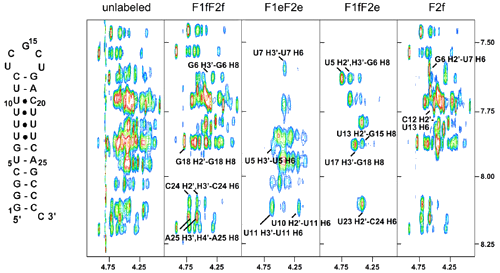
H/ACA RNA The pseudouridylation pocket of H/ACA snoRNPs H/ACA RNPs guide the modification of specific uridines in rRNA and snRNAs to pseudouridine, the most common modified nucleotide. They are essential for ribosomal RNA, snRNA, and telomerase RNA processing and metabolism. The box H/ACA RNPs are composed of four proteins including a pseudouridylase and an RNA containing a bipartite structure composed of two hairpins separated by conserved box H and ACA sequences. Each of the hairpins contains an internal loop that is complementary to the sequence surrounding the specific U targeted for modification, thus acting as ‘guides’. We have used the human U65 H/ACA snoRNA as our model system for structural studies. We determined the structure of the complex of the rRNA substrate sequence with the 3’ pseudouridylation pocket of U65hp. This was the first structure determination of a snoRNA with bound substrate, and provided complementary information to crystal structures of archeael H/ACA RNPs without bound substrate. Formation of the complex revealed that the substrate RNA can bind to the free H/ACA pseudouridylation pocket, thus opening up the “closed” internal loop, without the aid of protein co-factors. It also confirmed that the predicted secondary structure, with base pairs on either side of the pocket, and the target U and its 3’ nt unpaired at the top, is correct. The complex forms an unusual three-way junction, with co-axial stacking of the helix formed by the 5’ half of the substrate and the 3’ half of the pocket on the lower stem, and co-axial stacking of the helix formed by the 3’ half of the substrate and the 5’ half of the pocket on the upper stem. We have also compared the structures of the terminal loops of a scaRNAs and snoRNA to determine the sequence and structural requirements for Cajal body vs. nucleolar localization, respectively. Read more ...
In related work, we have investigated how pseudouridines in the P6.1 domain of human telomerase, identified by the Collins laboratory, affect telomerase RNA structure and activity. Read more ...
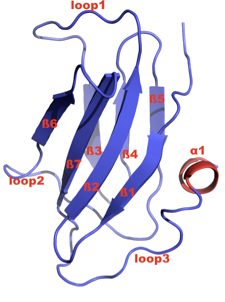
The H/ACA RNP assembly protein Shq1p Shq1p was identified by the Chanfreau laboratory to be an essential eukaryotic H/ACA snoRNP biogenesis and assembly factor. An orthologue has since been identified in humans. Shq1 is postulated to be involved in the early biogenesis steps of H/ACA snoRNP complexes. Depletion of Shq1 leads to a specific decrease in H/ACA snoRNA levels and to defects in ribosomal RNA processing. We are investigating the role of Shq1p in H/ACA RNP biogenesis using a combination of structural and functional studies. Shq1p contains two predicted domains: an N-terminal CS (named after CHORD-containing proteins and SGT1) or HSP20-like domain, and a C-terminal region of high sequence homology called the Shq1 domain. We recently determined the crystal structure and functional studies of the S. cerevisiae Shq1p CS domain. This is the first crystal structure determined in my laboratory. The structure consists of a compact anti-parallel β-sandwich fold that is composed of two β-sheets containing 4 and 3 β-strands, respectively, and a short α-helix. Based on the structure, we addressed two questions: first, whether the CS domain is important for Shq1p function and has a role in H/ACA snoRNP biogenesis, and second, whether it interacts with the Hsp90 molecular chaperone like the CS domains of the co-chaperones p23, Sba1 and Sgt1. Our results showed that the CS domain is required for the structural integrity and functions of Shq1p in vivo. There is a clear correlation between the structural properties and thermal stabilities of the isolated CS domains (wt and mutants) determined in vitro and the functional properties of these mutants observed in vivo. Several point mutations that destabilize the CS domain in vitro, as assessed by NMR and CD spectroscopy, generate growth defects, H/ACA snoRNA depletion phenotypes, and defects in rRNA processing, even in conditions when mutant Shq1p protein levels are normal. The Shq1p CS domain has the same fold as the CS domains of Hsp90 co-chaperones p23, Sba1, and Sgt1. Although CS domains are frequently found in co-chaperones of Hsp90 molecular chaperone, we found the Shq1p CS domain does not bind to yeast Hsp90 in vitro. These results show that the CS domain is essential for Shq1p function in H/ACA snoRNP biogenesis in vivo, possibly in an Hsp90-independent manner. Future studies are aimed at understanding how these Shq1 helps assembly the H/ACA RNPs. Read more ...
Recognition of snoRNP target sites by Rnt1 dsRBD Rnt1p, the only non-mitochondrial RNase III present in Saccharomyces cerevisiae, plays an essential role in the processing of ribosomal RNA (rRNA), small nuclear RNAs (snRNAs), and small nucleolar RNAs (snoRNAs) in budding yeast. Rnt1p is also important for mRNA quality control, cleaving intronic sequences of unspliced pre-mRNAs. A Rnt1 target site in the mRNA coding for the essential telomerase protein Est1 has been proposed to be important for maintenance of telomere length through regulation of Est1 expression. Although the RNAi pathway has been evolutionally lost in Saccharomyces cerevisiae, it has been found in the closely related budding yeast, e.g. Saccharomyces castellii and Kluyveromyces polysporus.
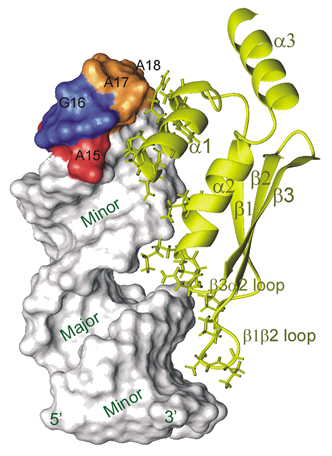
Rnt1 contains an endonuclease domain, a doublestrand RNA binding domain (dsRBD), and an N-terminal domain. Almost all Rnt1p substrates contain a hairpin capped by an AGNN tetraloop that is recognized by the Rnt1p dsRBD and dsRBD cleavage occurs 14-16 bp away from the tetraloop. We determined the structure a complex of Rnt1p dsRBD with a hairpin derived from the snR47 precursor, a snoRNA. This structure revealed that specificity of the dsRBD for AGNN tetraloop hairpins resides in structure specific recognition of a conserved tetraloop fold. The yeast Rnt1p dsRBD-AGAA hairpin complex structure was the first complex of any domain of a eukaryotic RNase III in complex with RNA to be determined.Although most Rnt1p substrates have AGNN tetraloops, snR48, identified as a substrate in a genome wide search, has an AAGU tetraloop. It was proposed that Rnt1p recognizes this substrate in a different way. In ongoing work, we are investigating how the dsRBD distinguishes dsRNA from substrate hairpins (specific vs non-specific binding) both in terms of structure and kinetics and the dynamics of the dsRBD in the absence and presence of RNA. Read more ...
Other Projects
Cation Binding to Nucleic Acids
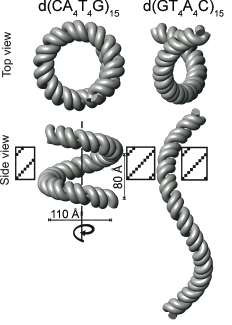
Cations play essential roles in nucleic acid structure, stability, folding, and catalysis. Nevertheless, the interactions between mono- and multivalent cations and nucleic acids are in many cases not well understood. We have been using both new (developed in our lab) and established NMR methods to study the interaction of cations with DNA and RNA. We identified specific monovalent cation binding sites on both DNA quadruplexes and on duplex DNA. One major goal is to understand how cations affect the structure of DNA A-tracts. Our laboratory was the first to show experimentally that cations could bind to specific sites within DNA A-tracts. We recently determined the solution structures of two DNA A-tracts with NH3+ counterions, and used these structures to create models of these DNAs in the context of 15 helical repeats. This NMR based structural modeling of d(CAAAATTTTG) and d(GTTTTAAAAC)15 surprisingly revealed that the former forms a left-handed superhelix with a diameter of 110Å and a pitch of ~80Å, similar to the DNA in the nucleosome, while the latter has only a gentle writhe and therefore appears nearly straight. Furthermore, the gel electrophoretic mobility of these DNA oligomers shows a monovalent cation dependence, providing further support that they play a fundamental role in DNA A-tract bending.
We are also studying the role of cations in RNA structure and ribozyme catalysis.
Methods Development for NMR Studies of Nucleic Acids
Many of our studies of nucleic acids have necessitated the development or optimization of NMR techniques for spectral assignment and structure determination of nucleic acids. We have an ongoing collaboration (since 1989) with Professor Vladimir Sklenar (Masaryk University, Brno, Czech Republic) to develop NMR methods for studying nucleic acids. Our current focus is on optimizing methods for studying RNA-protein complexes.
We have also collaborated with the laboratory of Stephen Grzesiek to study hydrogen bonding in nucleic acids, particularly DNA triplexes and quadruplexes, using measurements of scalar coupling constants between nuclei involved in hydrogen bonds.
In collaboration with Profs. Peter Qin and Wayne Hubbell, we have been developing and applying EPR site-directed spin labeling techniques to study the structure and dynamics of RNA, specifically the GAAA tetraloop receptor both free and bound to its receptor. This interaction is a frequently occurring motif that is important for long-range tertiary interactions in RNA. These methods will also be applicable to studies of larger RNAs.
Interactions of HHR23A with Cellular DNA Repair Proteins, the Ubiquitin/Proteosome Pathway, and HIV-1 Vpr
HHR23A, the human homologue of Rad23, is a multidomain protein containing an N-terminal Ubl (ubiquitin-like) domain, two UBA (ubiquitin associated) domains, and an XPC (Xeroderma pigmentosumcomplex C) protein-binding domain. Although HHR23A was first identified as a component of the DNA repair XPC complex, involved in nucleotide excision repair, recent studies also link it to the ubiquitin/proteosome pathway. The HIV-1 gene, vpr, encodes a 96 amino acid protein that is required for efficient infection of nondividing cells such as macrophages. Vpr induces cell cycle arrest at the G2/M checkpoint leading to subsequent apoptosis. Vpr interacts with a number of cellular proteins, includingHHR23A. Work by our collaborators in the I.S.Y. Chen lab at UCLA has provided support for the hypothesis that the Vpr/HHR23A/B interaction may be a key step in Vpr mediated cell cycle arrest. We determined the structure of the C-terminal UBA domain that interacts specifically with Vpr, and showed that mutation of a critical proline residue completely abolishes this interaction. Our current work on this project is focused on structure determination of other domains of HHR23A and their complexes with interacting proteins involved in DNA repair and ubiquitin/proteosome pathway. We have determined the structures of both of the UBA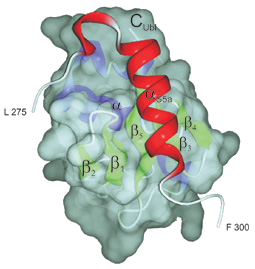 domains, the XPC-binding domain, and the Ubl domain. We have shown that the UBA domains interact with the b-sheet of ubiquitin via a conserved hydrophobic surface. We also recently reported the structure of a complex the Ubl domain with a UIM domain of the S5a subunit of the proteosome. This was the first structure of a UIM bound to a member of the ubiquitin family, and revealed the molecular determinants of the Ubl-proteasome interaction. The overall objective of these studies is to develop a comprehensive understanding of the structure/function relationships of HHR23A/B in cellular function, particularly as it relates to DNA repair, cell cycle, and ubiquitin/proteasome pathways. This understanding will, in turn, provide us with critical information needed to understand the role of the Vpr/HHR23A interaction in apoptosis.
domains, the XPC-binding domain, and the Ubl domain. We have shown that the UBA domains interact with the b-sheet of ubiquitin via a conserved hydrophobic surface. We also recently reported the structure of a complex the Ubl domain with a UIM domain of the S5a subunit of the proteosome. This was the first structure of a UIM bound to a member of the ubiquitin family, and revealed the molecular determinants of the Ubl-proteasome interaction. The overall objective of these studies is to develop a comprehensive understanding of the structure/function relationships of HHR23A/B in cellular function, particularly as it relates to DNA repair, cell cycle, and ubiquitin/proteasome pathways. This understanding will, in turn, provide us with critical information needed to understand the role of the Vpr/HHR23A interaction in apoptosis.
The Feigon Laboratory
Department of Chemistry and Biochemistry